

Abstract
Differences in Genomic Patterns Between African Americans and Whites with Acute Myeloid Leukemia
Abstract ID: 114273
Background:
In the United States, racial disparities in acute myeloid leukemia (AML) include African Americans (AAs) presenting at an earlier age compared to whites (a median of 48 vs 55 years) and having worse outcomes, with a 12% increased risk of death. Differences persist in studies demonstrating similar induction and post-remission treatment, treatment intensity, and follow-up for AA and whites, controlling for access to health care. To determine if disease biology plays a role, we studied the genomic profile of AA and white patients (pts) with AML.
Methods:
Pts diagnosed with and treated for AML at Cleveland Clinic between 2001-2013 with next generation target sequencing (NGS) available and pts from the The Cancer Genome Atlas (TCGA) with NGS available were included. Genomic panels of 60 gene mutations that are commonly mutated in AML and other myeloid malignancies were sequenced. Cytogenetic risk classification was per CALBG/Alliance criteria. Functional molecular groups were classified as follows: cohesion, splicing, DNA methylation, transcription, chromatin modifications, receptor/kinases, RAS pathway, RNA helicase, and tumor suppressor (see Table 1). Baseline characteristics and mutation incidence were compared between racial cohorts using Wilcoxon rank sum and chi-square tests. Survival analyses used log-rank and Cox proportional hazards regression.
Results:
Of 320 pts analyzed, 25 (7.8%) were AA and 295 (92.2%) were whites. AAs were younger than whites at diagnosis (median age of 59 [range 25-80] vs 65 years [21-100], P=.03) but had similar sex distribution (52% male vs 53.6%, P=1.0), median WBC (5.91 vs 5.29, P=.73) and blast percentage (47 vs 39, P=.30). Among AA pts, 18 (75%) had intermediate-risk cytogenetics compared to 222 whites (76.6%); 6 (25%) had unfavorable cytogenetics compared to 68 whites (23.4%, P=1.0). Overall survival in AA vs whites was similar at a median of 10.6 (range .13-63) vs 14.6 months (range .03-114) (P = .50), and when controlling for cytogenetics and age (HR 1.05, 95% CI 0.60-1.85, P =.87).
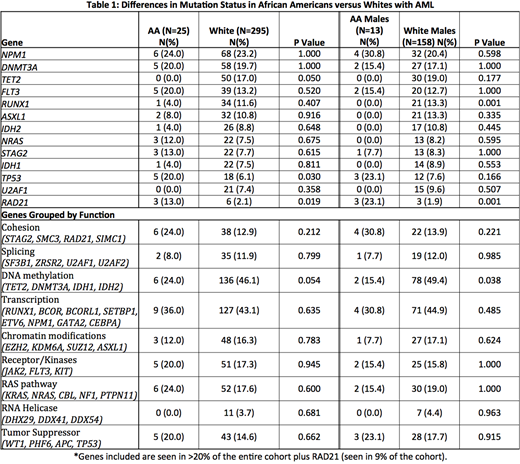
Overall, the most frequently mutated genes were NPM1 (seen in 74% of all pts), DNMT3A (63%), TET2 (50%), FLT3 (44%), RUNX1 (35%), ASXL1 (34%), IDH2 (27%), NRAS (25%), STAG2 (25%), IDH1 (23%), TP53 (23%), U2AF1 (21%). The median number of mutations per sample was 2 (range 0 - 15) and there was no difference between AA and whites (median 2 [range 0-6] vs 2 [0-15], P= 0.35). AA were more likely to have mutations in RAD21 (13 vs 2%, P=.019) and TP53 (20 vs 6%, P=.03), and less likely to present with TET2 (0 vs 17%, P=0.050). When mutations were grouped by function, mutations in genes for methylation were less common in AA (n=2, 15.4%) vs whites (n=78, 49.4%) (P=.038). No other differences were seen between groups when mutations were grouped by function, see Table 1.
Stratified analysis by sex, comparing AA males to white males, revealed that the prevalence of RAD21 was higher in AA males (23 vs 2%, P<.001). TP53 was mutated in 23% of AA males vs 7.6% of white males (P=.166).
Survival analyses, adjusting for age, estimated a 10-fold increase in death rate for AA with a RAD21 mutation compared to whites with a RAD21 mutation (HR 0.1, 95% CI 0.01-1.82, P=.12). Mutations in TP53 were associated with a non-significant survival difference in AA vs whites (HR 0.68, 95% CI 0.24-1.90, P=.46)
Conclusion:
AA pts with AML are younger at diagnosis compared to whites and more likely to have TP53 (consistent with previous studies showing higher rates of complex cytogenetics) and RAD21 mutations, but less likely to have TET2 mutations and methylation gene mutations. Some molecular abnormalities may be sex-specific. RAD21 is more common in AA males than white males and may contribute to excess death rates. These findings may help explain worse survival in AA and may suggest differential therapeutic efficacy.
Nazha:MEI: Consultancy. Carraway:Agios: Consultancy, Speakers Bureau; Jazz: Speakers Bureau; Balaxa: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Celgene: Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; FibroGen: Consultancy; Novartis: Speakers Bureau; Amgen: Membership on an entity's Board of Directors or advisory committees. Gerds:Celgene: Consultancy; Apexx Oncology: Consultancy; Incyte: Consultancy; CTI Biopharma: Consultancy. Advani:Amgen: Research Funding; Pfizer: Honoraria, Research Funding; Glycomimetics: Consultancy; Novartis: Consultancy. Maciejewski:Ra Pharmaceuticals, Inc: Consultancy; Apellis Pharmaceuticals: Consultancy; Apellis Pharmaceuticals: Consultancy; Alexion Pharmaceuticals, Inc.: Consultancy, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Ra Pharmaceuticals, Inc: Consultancy; Alexion Pharmaceuticals, Inc.: Consultancy, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau. Sekeres:Celgene: Membership on an entity's Board of Directors or advisory committees; Opsona: Membership on an entity's Board of Directors or advisory committees; Celgene: Membership on an entity's Board of Directors or advisory committees; Opsona: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal